Resources
About Us
Advanced Biologics Manufacturing Market by Biologic Type, Workflow Stage Product and Service, Application, End User (Biopharmaceutical Companies, CDMOs and CMOs, Academic and Research Institutes) - Global Forecast to 2036
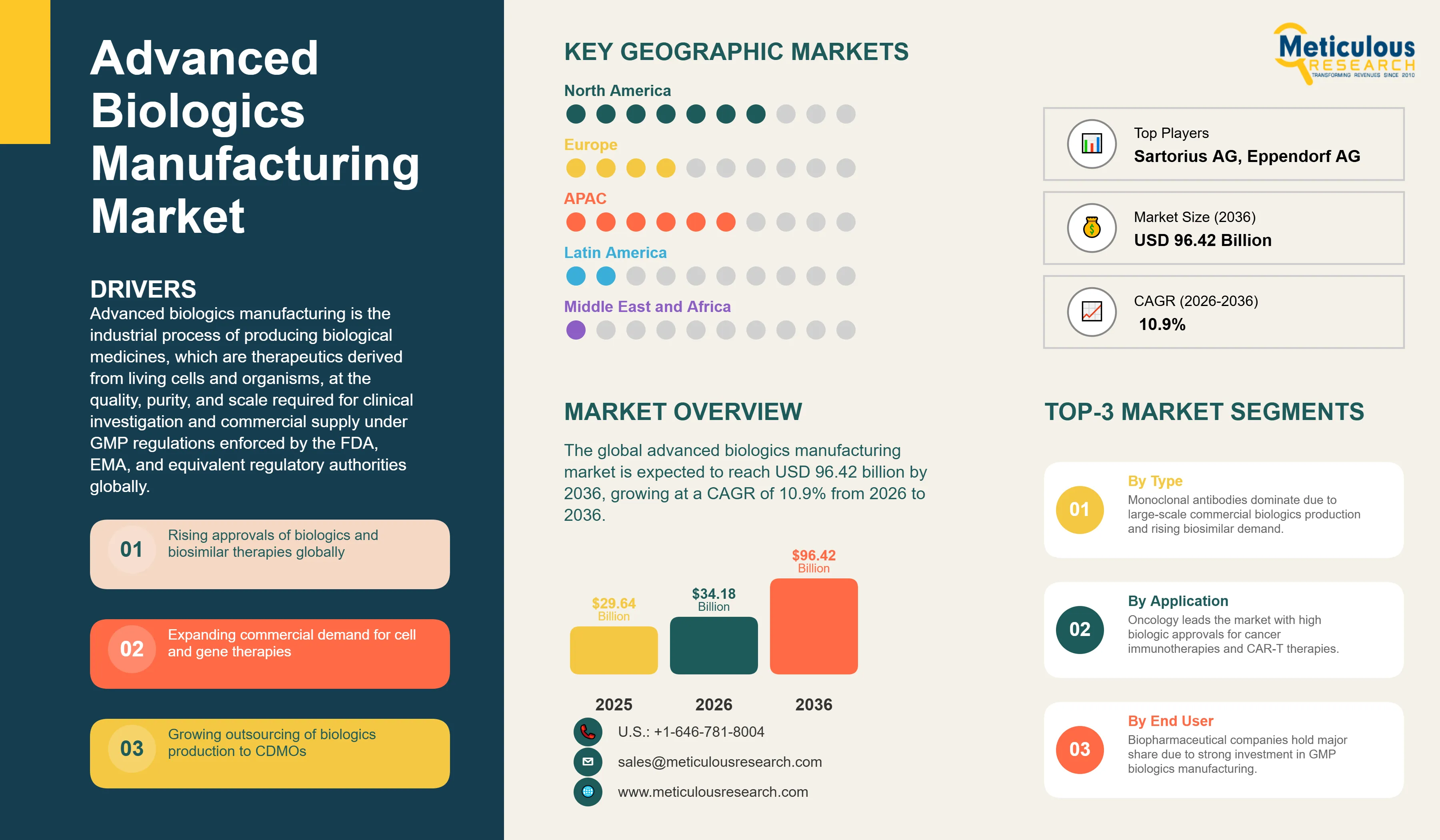
Report ID: MRHC - 1041985 Pages: 290 May-2026 Formats*: PDF Category: Healthcare Delivery: 24 to 72 Hours Download Free Sample ReportThe global advanced biologics manufacturing market was valued at USD 29.64 billion in 2025. This market is expected to reach USD 96.42 billion by 2036 from USD 34.18 billion in 2026, growing at a CAGR of 10.9% from 2026 to 2036.
The growth of this market is driven by the sustained expansion of the biologics pipeline, the increasing commercial scale of approved biologic therapies, and the structural investment in manufacturing infrastructure required to deliver complex biological molecules at global commercial volumes. Advanced biologics manufacturing encompasses the full range of GMP-compliant production processes used to manufacture monoclonal antibodies, recombinant proteins, vaccines, cell therapies, gene therapies, and other biological products derived from living organisms. These manufacturing processes require specialized facilities, cell culture systems, purification platforms, and quality control infrastructure that collectively represent the most technically demanding and capital-intensive segment of the pharmaceutical manufacturing industry.
According to the FDA's Center for Biologics Evaluation and Research CY 2024 Report from the Director, CBER approved 17 biologics license applications and 26 biologics license application supplements in 2024 alone, in addition to 24 biological device applications. In 2023, CBER approved 20 biologics license applications and 17 BLA supplements. As of March 2026, the FDA has approved 82 biosimilar products through the abbreviated licensure pathway established by the Biologics Price Competition and Innovation Act of 2009. These approvals provide increased access to treatments for conditions like cancer and diabetes, with FDA-approved biosimilars saving the U.S. healthcare system $23.6 billion since 2015. Based on reports from the FDA's Office of Therapeutic Products (OTP) in 2023–2024, there were indeed over 2,500 active investigational new drug applications (INDs) for cellular and gene therapies (CGTs). Of that total, approximately 1,300 active INDs were specifically focused on gene therapies, highlighting a robust, fast-growing pipeline. Each of these approved products and investigational programs requires GMP-compliant manufacturing at one or more production stages, creating the sustained and compounding demand for advanced biologics manufacturing capacity, technology, and services that defines the commercial market.
Advanced biologics manufacturing is the industrial process of producing biological medicines, which are therapeutics derived from living cells and organisms, at the quality, purity, and scale required for clinical investigation and commercial supply under GMP regulations enforced by the FDA, EMA, and equivalent regulatory authorities globally. The product scope of the market encompasses monoclonal antibodies and antibody-derived formats including bispecific antibodies, antibody-drug conjugates, and Fc-fusion proteins; recombinant proteins including insulin, erythropoietin, clotting factors, and enzyme replacement therapies; prophylactic and therapeutic vaccines produced in cell culture, egg-based, or mRNA manufacturing systems; cell therapies including autologous and allogeneic CAR-T, NK cell, and tumor-infiltrating lymphocyte products; gene therapies including AAV and lentiviral vector-based in vivo and ex vivo gene delivery products; and emerging modalities including mRNA therapeutics and oncolytic viruses.
The manufacturing of each biologic product class requires a distinct set of production platforms, upstream cell culture systems, downstream purification processes, and analytical characterization methods. Monoclonal antibodies are produced at commercial scale primarily in Chinese hamster ovary cell cultures in stirred-tank bioreactors of up to 25,000 liters, with protein A affinity chromatography as the primary capture step and a series of polishing and viral clearance steps completing the downstream process. Recombinant proteins are manufactured in CHO, E. coli, and yeast expression systems depending on the glycosylation requirements of the specific molecule. Vaccines are produced using a diverse range of platforms including inactivated virus produced in embryonated eggs or cell culture, live attenuated virus, recombinant protein antigens, and lipid nanoparticle-formulated mRNA. Cell therapies require personalized or batch manufacturing of living cellular products using closed automated cell processing systems, expansion bioreactors, and cryopreservation and distribution infrastructure that distinguishes them fundamentally from conventional biologics manufacturing. Gene therapies use viral vector production processes as described in the broader cell and gene therapy manufacturing literature.
The competitive landscape of the advanced biologics manufacturing market is organized around two primary commercial models. CDMOs including Lonza, Samsung Biologics, Fujifilm Diosynth Biotechnologies, WuXi Biologics, Catalent, and AGC Biologics provide contract manufacturing services under agreements with biopharmaceutical companies. Technology and equipment providers including Thermo Fisher Scientific, Cytiva, Sartorius, Merck KGaA, Repligen, Pall Corporation, Bio-Rad, and Eppendorf supply the bioreactors, filtration systems, chromatography platforms, cell culture media, and analytical instruments used across the manufacturing workflow by both CDMOs and in-house manufacturing operations. The market also includes specialized contract service providers for process development, cell line development, analytical method development, and regulatory affairs support that form the extended service ecosystem around core GMP manufacturing.
Transition to Single-use Manufacturing Systems Across the Industry
The adoption of single-use bioreactor systems and single-use downstream processing components as the preferred infrastructure for new biologics manufacturing capacity is the most broadly impactful technology transition in advanced biologics manufacturing. Single-use systems use pre-sterilized, gamma-irradiated plastic contact surfaces for bioreactors, filtration assemblies, tubing, and fluid management components that are discarded after each production batch rather than cleaned, sterilized, and revalidated as conventional stainless steel equipment is. This shift eliminates the cleaning validation and cross-contamination risk management requirements that add cost, time, and regulatory complexity to stainless steel manufacturing operations. The FDA's 2024 guidance on CMC flexibilities for cell and gene therapy products explicitly acknowledges single-use systems as a manufacturing approach appropriate for CGT product manufacturing, reflecting the agency's recognition of single-use technology as an accepted platform across the biologics manufacturing regulatory framework.
The commercial adoption of single-use systems has been driven primarily by the CDMO sector, where the flexibility to switch between different client products without the downtime and cost of stainless steel cleaning and revalidation cycles provides a significant operational advantage. Sartorius, Cytiva, Thermo Fisher Scientific, and Pall Corporation have developed comprehensive single-use bioprocessing portfolios that span bioreactor bags and vessels, tangential flow filtration cassettes, depth filtration devices, and fluid management assemblies for the complete upstream and downstream biologics manufacturing workflow. The single-use market has expanded from early adoption in clinical-scale manufacturing to broader deployment at commercial scales of 500 liters and above, with modular single-use facilities now being specified for commercial biologics manufacturing by leading CDMOs and biopharmaceutical companies.
FDA and EMA Regulatory Frameworks Evolving to Support Advanced Therapy Manufacturing
The regulatory frameworks governing advanced biologics manufacturing, particularly for cell and gene therapy products, are evolving rapidly as the FDA and EMA develop guidance specific to the manufacturing challenges of these novel modalities. The FDA's CBER has issued a substantial body of guidance on CGT manufacturing between 2019 and 2025, addressing CMC requirements for gene therapy INDs, manufacturing changes and comparability for CGT products, potency assurance for cellular and gene therapy products, and the use of human and animal-derived materials in CGT manufacture. According to the FDA CBER CY 2024 Director's Report, CBER continued to advance its CMC Development and Readiness Pilot program in 2024, designed to facilitate CMC development for products with expedited clinical development timelines and support manufacturers in building commercial-ready manufacturing processes earlier in clinical development. The FDA's CoGenT Global Pilot, a collaboration with international regulatory partners, advances global harmonization of CGT regulatory requirements, reducing the burden of parallel regulatory submissions for manufacturers seeking approval in multiple markets. These regulatory program developments are directly relevant to biologics manufacturers because they define the GMP compliance requirements, analytical testing standards, and manufacturing change management processes that manufacturers must build into their operations, equipment specifications, and quality management systems.
|
Parameter |
Details |
|
Market Size by 2036 |
USD 96.42 Billion |
|
Market Size in 2026 |
USD 34.18 Billion |
|
Market Size in 2025 |
USD 29.64 Billion |
|
Market Growth Rate (2026-2036) |
CAGR of 10.9% |
|
Dominating Region |
North America |
|
Fastest Growing Region |
Asia-Pacific |
|
Base Year |
2025 |
|
Forecast Period |
2026 to 2036 |
|
Segments Covered |
Biologic Type, Workflow Stage, Product and Service, Technology Platform, Application, End User, and Region |
|
Regions Covered |
North America, Europe, Asia-Pacific, Latin America, and Middle East & Africa |
Driver: Rising Volume of FDA and EMA Biologic Approvals Driving Commercial Manufacturing Demand
The sustained high rate of biologic product approvals by the FDA and EMA is the primary near-term driver of commercial-scale advanced biologics manufacturing demand. Each newly approved biologic product requires the transition from clinical-scale GMP manufacturing to commercial-scale production, involving the scale-up of upstream cell culture processes to larger bioreactor volumes, the validation of commercial downstream purification processes, and the qualification of commercial fill and finish operations. According to the FDA CBER CY 2024 Report from the Director, CBER approved 17 biologics license applications in 2024 and approved 20 BLAs in 2023, confirming the sustained pace of biologic approvals flowing into the commercial manufacturing pipeline. According to FDA CDER records, additional biologic approvals are processed through CDER's Center, including monoclonal antibodies and recombinant proteins that are regulated as drugs rather than CBER biologics, with CDER's Office of Oncologic Diseases alone approving 17 novel drugs and biologics for cancer treatment in 2024 and completing 32 additional approval decisions for expanded indications. The cumulative commercial manufacturing demand generated by this approval volume, combined with the growing commercial sales of existing blockbuster biologics requiring continuous large-scale production, creates the foundational demand driver for the advanced biologics manufacturing market across all segments from upstream equipment to downstream consumables and CDMO services.
Opportunity: Continuous Bioprocessing and Integrated Manufacturing Platform Development
The development and adoption of continuous bioprocessing approaches, which replace the batch-by-batch production model of conventional biologics manufacturing with a continuously operating integrated process, represents a significant opportunity to improve manufacturing productivity, reduce facility footprint, and lower the cost of goods for biologic medicines. In conventional batch biologics manufacturing, the bioreactor is inoculated, cells are grown to peak density, the harvest is taken at a single time point, and the bioreactor is then cleaned and prepared for the next batch. Continuous upstream perfusion processes, in which fresh medium is continuously fed to the bioreactor and spent medium plus product are continuously removed, maintain cells at high density for extended periods and generate substantially higher volumetric productivity per liter of bioreactor capacity than batch or fed-batch processes. The FDA's Process Analytical Technology guidance framework and the ICH Q13 guideline on continuous manufacturing of small and large molecule drug substances, finalized in 2023, provide the regulatory framework that is enabling manufacturers to implement continuous bioprocessing with regulatory predictability. The FDA's CBER has acknowledged in its guidance program that continuous manufacturing approaches are applicable to biologics and has committed to providing regulatory clarity for sponsors seeking to implement integrated continuous bioprocessing for advanced biologic products. Adoption of continuous manufacturing is expected to grow significantly during the forecast period, with Sartorius, Cytiva, and Repligen all offering perfusion bioreactor and continuous downstream processing equipment designed for integrated manufacturing workflows.
Why Do Monoclonal Antibodies Lead the Market?
In 2026, the monoclonal antibodies segment is expected to hold the largest share of the advanced biologics manufacturing market. Monoclonal antibodies are the most commercially mature and highest-revenue category of biologic medicines globally. Multiple approved mAb products generate annual revenues exceeding USD 10 billion per year individually, including Merck's pembrolizumab, Bristol Myers Squibb's nivolumab, AbbVie's adalimumab, and Roche's trastuzumab and bevacizumab, creating sustained high-volume commercial manufacturing demand that constitutes the majority of total global bioreactor capacity utilization. The manufacturing process for mAbs is well established and highly optimized, with CHO cell culture in stirred-tank bioreactors, protein A affinity capture, and a series of chromatography and filtration polishing steps representing the industry-standard platform process. The biosimilar market is also driving mAb manufacturing demand growth, as multiple biosimilars have been approved for major reference mAbs. According to the FDA, biosimilar products have been approved for 15 different reference biologics, the majority of which are monoclonal antibodies, and FDA-approved biosimilars have collectively generated approximately 700 million patient-days of therapy, with 344 million days representing treatment patients may not otherwise have received. Each biosimilar approval requires a separate GMP manufacturing operation, multiplying the number of manufacturing sites and production runs required globally to serve the mAb therapeutic category.
However, the cell therapies segment is expected to witness the fastest growth during the forecast period. The commercial expansion of approved CAR-T cell therapies and the large pipeline of next-generation cell therapy programs advancing through clinical development with FDA CBER oversight are driving strong manufacturing demand growth. According to FDA CBER, approximately 2,500 active INDs for cell and gene therapies were on file with the Office of Therapeutic Products in 2023 and 2024. The FDA's CY 2024 Director's Report confirmed that CBER approved an efficacy supplement for axicabtagene ciloleucel and a safety supplement for brexucabtagene autoleucel in 2024, reflecting the ongoing regulatory activity around approved commercial CAR-T products. Each commercial autologous CAR-T treatment requires a dedicated patient-specific manufacturing run, and the growing number of approved CAR-T products in commercial use is generating recurring, patient-volume-driven manufacturing demand that grows with commercial adoption.
How Does Upstream Processing Lead the Market?
In 2026, the upstream processing segment is expected to hold the largest share of the advanced biologics manufacturing market. Upstream processing encompasses cell culture systems, bioreactors, cell culture media, transfection systems, and associated consumables used to produce the biological product within the expression system. This stage represents the largest single capital and recurring cost component of the biologics manufacturing workflow, with large-scale stainless steel bioreactors and single-use bioreactor systems constituting the most significant capital equipment investment in a biologics manufacturing facility. The cell culture media and supplement market generates high recurring revenue as it is consumed with every production batch. Thermo Fisher Scientific, Sartorius, Cytiva, and Merck KGaA are the primary suppliers of upstream bioprocessing equipment and consumables, and their combined market positions in this segment make upstream the largest revenue category within the biologics manufacturing value chain.
However, the downstream processing segment is expected to witness the fastest growth during the forecast period. As biologics manufacturing processes are intensified and upstream volumetric productivity increases through process improvements, the volume of material requiring downstream purification per facility per year increases proportionally, driving demand for higher-throughput downstream processing equipment and consumables. The growing complexity of advanced biologic products including bispecific antibodies, ADCs, cell therapy manufacturing intermediates, and viral vectors places greater demands on downstream purification selectivity and efficiency than conventional mAb manufacturing, driving investment in new chromatography resin technologies, advanced filtration devices, and continuous downstream processing systems. Repligen and Pall Corporation in particular are active in developing next-generation downstream processing equipment specifically designed for the complex purification challenges of advanced biologic products.
Why Do Services Lead the Market?
In 2026, the services segment is expected to hold the largest share of the advanced biologics manufacturing market. Contract manufacturing services provided by CDMOs represent the largest single revenue category in the market because the majority of biopharmaceutical companies, particularly those in clinical development stages, rely on CDMO partners for their GMP biologics manufacturing needs. Establishing in-house biologics manufacturing capability requires capital investment of several hundred million dollars for a purpose-built GMP facility, regulatory validation timelines of two to five years, and a specialized workforce that is in chronic short supply globally. The CDMO services model captures premium revenue over the underlying manufacturing cost by providing clients with GMP-qualified facilities, validated processes, regulatory expertise, and supply chain risk management that represent significant value in supporting clinical programs toward regulatory approval. According to FDA CBER, the CMC Development and Readiness Pilot program, which CBER operated in collaboration with CDER in 2024, was designed specifically to facilitate earlier development of commercial-ready manufacturing processes for products with expedited development timelines, confirming the regulatory agency's recognition that manufacturing readiness is a critical bottleneck in the biologics development pathway that CDMO partnerships are uniquely positioned to address.
However, the consumables and reagents segment is expected to witness the fastest growth during the forecast period. As the volume of biologics manufacturing activity grows with product approvals and clinical pipeline expansion, the recurring demand for single-use bioreactor bags, cell culture media, chromatography resins, filtration devices, and kits and assays grows proportionally. Unlike capital equipment, which is purchased infrequently, consumables are replaced with every production batch and every analytical testing cycle, creating a high-frequency recurring revenue stream that scales directly with manufacturing volume. Sartorius, Cytiva, Thermo Fisher Scientific, and Merck KGaA have built their commercial strategies around this recurring consumable revenue model, with single-use bioreactor platforms in particular generating a captive stream of bag and tubing assembly revenue from every campaign conducted on the installed platform base.
Why Do Single-use Technologies Lead the Market?
In 2026, the single-use technologies segment is expected to hold the largest share of the advanced biologics manufacturing market by technology platform. Single-use systems have become the dominant infrastructure standard for new biologics manufacturing capacity investment, particularly in CDMO operations and clinical-stage biologics manufacturing. The FDA's regulatory framework for biologics manufacturing, including its guidance on manufacturing changes and comparability and its 2024 guidance on CMC flexibilities for cell and gene therapy products, accommodates single-use systems as a validated manufacturing approach for GMP biologics production, providing manufacturers with the regulatory certainty needed to commit to single-use infrastructure at commercial scale. The combination of eliminated cleaning validation burden, reduced cross-contamination risk, faster facility setup timelines, and operational flexibility to switch between product campaigns makes single-use systems the preferred choice for new capacity investment across the CDMO sector and increasingly for commercial in-house manufacturing as well. Sartorius Biostat STR, Cytiva Xcellerex, Thermo Fisher Scientific HyPerforma, and Pall Allegro systems are the leading commercial single-use bioreactor platforms.
However, the automation and digital bioprocessing segment is expected to witness the fastest growth during the forecast period. The integration of automated bioprocess control, process analytical technology sensors, and digital data management platforms into biologics manufacturing operations is accelerating as manufacturers seek to reduce variability, improve batch consistency, and satisfy the FDA's Process Analytical Technology guidance framework which encourages real-time process monitoring and control. The FDA's ICH Q13 guideline on continuous manufacturing, finalized in 2023 and applicable to biological products, provides the regulatory framework for continuous bioprocessing automation, creating a pathway for manufacturers to implement integrated automated continuous manufacturing with regulatory predictability. Cytiva's KrosFlo and AKTA systems and Sartorius's ambr and biostat platforms are developing integrated automation capabilities that bring digital process control to both upstream and downstream biologics manufacturing workflows.
Why Does Oncology Lead the Application Market?
In 2026, the oncology segment is expected to hold the largest share of the advanced biologics manufacturing market. Oncology is the largest therapeutic area for biologic drug development and approval, encompassing monoclonal antibody checkpoint inhibitors, antibody-drug conjugates, CAR-T cell therapies, cancer vaccines, bispecific antibodies, and recombinant protein growth factorsAccording to the FDA Oncology Regulatory Review 2024, the Office of Oncologic Diseases (OOD) approved 17 novel cancer therapies, including treatments for solid tumors and blood cancers. In addition to these 17 new approvals, the FDA completed 32 supplementary approvals to expand the use or population for existing therapies. The FDA's CBER also approved supplemental applications for axicabtagene ciloleucel and brexucabtagene autoleucel, two approved commercial CAR-T products, in 2024. In addition, CDRH in collaboration with FDA's Oncology Center of Excellence authorized 76 oncology devices in 2024. The breadth and volume of oncology biologic approvals, combined with the large commercial manufacturing volumes required by blockbuster checkpoint inhibitor therapies, makes oncology the dominant application segment in the biologics manufacturing market.
However, the rare diseases segment is expected to witness the fastest growth during the forecast period. The FDA CBER has placed rare disease therapeutics at the center of its advanced therapy development and approval strategy. According to the FDA CBER CY 2024 Director's Report, CBER approved multiple products for rare diseases in 2024 and advanced its Support for clinical Trials Advancing Rare disease Therapeutics Pilot Program, with selected participants notified of their acceptance on May 29, 2024. CBER and CDER jointly announced the FDA Rare Disease Innovation Hub in 2024, a cross-agency initiative to enhance the development of treatments for rare diseases with a focus on products for smaller populations and diseases with variable natural histories. The Rare Disease Endpoint Advancement Pilot Program continued to support novel endpoint development for rare disease drugs and biologics in 2024. The growing pipeline of gene therapies, enzyme replacement therapies, and cell therapies targeting rare genetic disorders is driving significant manufacturing demand growth in the rare disease application segment.
Why Do Biopharmaceutical Companies Lead the End User Market?
In 2026, the biopharmaceutical companies segment is expected to hold the largest share of the advanced biologics manufacturing market. Innovator biopharmaceutical companies including Roche, Pfizer, Johnson and Johnson, AbbVie, Amgen, Biogen, Regeneron, and Novo Nordisk own the approved biologic products that generate the majority of commercial manufacturing demand globally. These companies maintain large in-house manufacturing operations for their approved commercial products and are the primary buyers of advanced biologics manufacturing equipment, consumables, and process development services. They are also the primary entities that seek BLA approval from the FDA for new biologic products and that fund the clinical manufacturing programs at CDMOs for their pipeline assets. According to FDA CDER, biologics manufacturers must submit chemistry, manufacturing, and controls information in their BLA applications and post-approval manufacturing change supplements, creating ongoing regulatory compliance requirements that drive sustained investment in manufacturing quality systems, process validation, and analytical capabilities.
However, the CDMOs and CMOs segment is expected to witness the fastest growth during the forecast period. The structural outsourcing trend in biologics manufacturing is accelerating as biopharmaceutical companies seek to reduce capital investment in manufacturing infrastructure and access the manufacturing expertise, regulatory track record, and multi-product operational experience that leading CDMOs provide. Samsung Biologics has expanded its total bioreactor capacity to among the largest globally, WuXi Biologics continues to expand its multi-site manufacturing network across Asia, Europe, and the United States, and Fujifilm Diosynth Biotechnologies is expanding capacity specifically for advanced modalities including viral vectors and cell therapy manufacturing. The FDA's CMC Development and Readiness Pilot program, noted in CBER's 2023 and 2024 Director's Reports, is specifically designed to support earlier manufacturing readiness, which CDMO partnerships are positioned to facilitate through their established GMP infrastructure and validated process platforms.
How is North America Maintaining Market Leadership?
In 2026, North America is expected to hold the largest share of the global advanced biologics manufacturing market. The United States is the primary market, driven by the world's largest concentration of innovator biopharmaceutical companies, the highest volume of biologics regulatory approvals globally, and the largest installed base of GMP biologics manufacturing capacity.
According to the FDA CBER CY 2024 Director's Report, CBER approved 17 biologics license applications in 2024, and according to the FDA CDER Oncology Regulatory Review 2024, CDER's Office of Oncologic Diseases approved 17 novel oncology drugs and biologics and completed 32 additional approval decisions in 2024. This approval volume is without parallel globally and directly drives commercial manufacturing demand at U.S.-based and CDMO facilities serving U.S.-licensed products. The FDA's active program of guidance issuance for advanced therapy manufacturing, including the 2024 guidance on CMC flexibilities for cell and gene therapy products, the CMC Development and Readiness Pilot, and the CoGenT Global Pilot for international regulatory harmonization, confirms the U.S. regulatory environment's commitment to supporting manufacturing innovation and capacity expansion for advanced biologics. Canada contributes to regional demand through established biotechnology clusters in Toronto, Montreal, and Vancouver and the National Research Council's biologics manufacturing programs.
Which Factors Drive Asia-Pacific's Rapid Growth?
Asia-Pacific is expected to witness the highest growth rate in the advanced biologics manufacturing market during the forecast period. This growth is driven by the rapid expansion of CDMO capacity in China, South Korea, Singapore, and India, growing domestic biologics development pipelines, and government-supported biopharmaceutical manufacturing investment across the region.
China is the largest individual market in Asia-Pacific for advanced biologics manufacturing. WuXi Biologics, a leading Chinese biologics CDMO, has been expanding its manufacturing network across China and internationally, building new GMP bioreactor capacity to serve the growing global demand for biologics contract manufacturing. Chinese domestic biopharmaceutical companies are developing an expanding biologics pipeline, with an increasing number of BLA submissions to both Chinese regulatory authorities and to the FDA and EMA for global registration. South Korea is home to Samsung Biologics, which has built one of the world's largest single-site biologics manufacturing complexes in Incheon, with total bioreactor capacity designed to serve large-volume commercial mAb manufacturing programs. Singapore is positioned as the regional hub for high-value biologics manufacturing, with Lonza, Thermo Fisher Scientific, and AGC Biologics operating or expanding GMP biologics manufacturing facilities supported by the Singapore Economic Development Board. India's biologics manufacturing sector, anchored by the Serum Institute of India, Biocon Biologics, and Dr. Reddy's Laboratories, is expanding its advanced biologics manufacturing capability beyond vaccine production into recombinant proteins, biosimilars, and the early stages of cell and gene therapy manufacturing capacity development.
Some of the key companies operating in the global advanced biologics manufacturing market are Thermo Fisher Scientific Inc., Danaher Corporation (Cytiva), Sartorius AG, Merck KGaA, Lonza Group AG, Fujifilm Diosynth Biotechnologies, Samsung Biologics, WuXi Biologics, Catalent, Inc., AGC Biologics, Charles River Laboratories International Inc., Bio-Rad Laboratories, Inc., Eppendorf AG, Repligen Corporation, and Pall Corporation.
The global advanced biologics manufacturing market is expected to grow from USD 34.18 billion in 2026 to USD 96.42 billion by 2036.
The global advanced biologics manufacturing market is projected to grow at a CAGR of 10.9% from 2026 to 2036.
The monoclonal antibodies segment is expected to dominate the overall market in 2026, supported by the large commercial manufacturing volumes of approved blockbuster mAb therapies and the growing biosimilar mAb manufacturing base. However, the cell therapies segment is expected to witness the fastest CAGR, driven by the commercial expansion of FDA-approved CAR-T products and the approximately 2,500 active CGT INDs on file with FDA CBER creating a large and growing pipeline of next-generation cell therapy programs requiring GMP manufacturing.
The single-use technologies segment is expected to dominate the overall market in 2026. However, the automation and digital bioprocessing segment is expected to witness the fastest CAGR, driven by FDA's Process Analytical Technology framework encouraging real-time process monitoring and the ICH Q13 guideline on continuous manufacturing providing regulatory clarity for integrated automated bioprocessing implementation
The oncology segment is expected to dominate the overall market in 2026, anchored by the FDA OOD's 17 novel oncology drug and biologic approvals and 32 additional approval decisions in 2024. However, the rare diseases segment is expected to witness the fastest CAGR, driven by the FDA's Rare Disease Innovation Hub, the START Pilot Program, and the growing gene therapy pipeline targeting rare genetic conditions.
North America is expected to lead the global market in 2026, supported by the FDA's sustained high volume of BLA approvals driving commercial manufacturing demand. However, Asia-Pacific is expected to witness the fastest CAGR, driven by rapid CDMO capacity expansion in China, South Korea, and Singapore.
The major players are Thermo Fisher Scientific, Cytiva (Danaher), Sartorius, Merck KGaA, Lonza Group, Fujifilm Diosynth Biotechnologies, Samsung Biologics, WuXi Biologics, Catalent, AGC Biologics, Charles River Laboratories, Bio-Rad Laboratories, Eppendorf, Repligen Corporation, and Pall Corporation.
























Published Date: May-2024
Published Date: Feb-2024
Please enter your corporate email id here to view sample report.
Subscribe to get the latest industry updates